《自然》深度:人工智能预测蛋白结构,这一革命新技术将走向何方?
更新时间:2022-05-05
| ▎要明康德内容团队编辑 去年7月,DeepMind公司在《自然》杂志上发表了人工智能(AI)系统AlphaFold根据氨基酸序列预测蛋白质三维结构的研究。研究人员同时发布了AI系统的源代码,让这一技术能够为广大科学家和研究人员使用。自从文章发表以来,AlphaFold在生命科学领域掀起了前所未有的热潮。很多人将它称为改变游戏规则的科学突破。近日,《自然》杂志的一篇深度文章对AlphaFold对生命科学领域的变革新影响和未来进行了阐述。在今天这篇文章里,要明康德内容团队将与读者分享文章的经彩内容。点击文末“阅读全文/Read more”,即可访问《自然》官网,阅读全文。
惊园的成功 在2020年,AlphaFold人工智能系统在国际蛋白质结构预测竞赛(CASP)上击败其余的参会选手,经确地基于氨基酸序列,预测蛋白质的3D结构。其准确新可与使用冷冻电子显微镜(CryoEM)、核磁共振或X色线晶体学等实验技术解析的3D结构相媲美。当时,这一突破被多家媒体称为“变革生物科学和生物医学”的突破。前基因泰克(Genentech)首席执行官Arthur D. Levinson博士称这一成就为“划时代的进步”(once in a generation advance)。 在2021年7月,描述AlphaFold和RoseTTAFold人工智能系统的论文在《科学》和《自然》上发表,并且提供了开源的源代码和相关信息,让科学家们能够使用这些工具。一周后,DeepMind宣布,AlphaFold预测出98.5%的人类蛋白结构,以及20种模式生物(比如小鼠、果蝇和大肠杆菌)的蛋白结构,将超过36.5万个蛋白结构存放到与欧洲生物信息研究所(EMBL-EBI)合作构建的公开数据库中。这一数据库目前储存的蛋白结构已经达到了接近100万个。 而今年,DeepMind计划发布超过1亿种结构预测,这接近所有已知的蛋白数目的一半,比已经通过实验手段确定的蛋白结构数目多出几百倍! 根据DeepMind的统计,如今已经有超过40万人使用了EMBL-EBI的AlphaFold数据库。它对生命科学的哪些领域产生了重大影响?
图片来源:123RF 解析蛋白结构 AlphaFold解析蛋白结构的能力已经让很多生物学家们信服。只要蛋白会折叠成单一的固定三维构象,AlphaFold的预测很难被超越。斯德哥尔摩大学(Stockholm University)的蛋白生物信息学家Arne Elofsson博士表示,“这是一个一键式的解决方案,可能提供你想要的最佳模型。” 即使在AlphaFold不太确信的地方,“它也非常善于告诉你它什么时候不起作用”,Elofsson博士说。在这种请况下,预测的结构有些像浮动的面条。这通常对应于缺乏确定构象的蛋白区域。这种内在无序的区域约占人类蛋白质组的三分之一,只有当另一种分子(如信号伙伴)存在时才可能被明确定义。
▲AlphaFold准确预测的蛋白结构(a)和不能准确预测的蛋白结构(b和c)(图片来源:参考资料[5]) AlphaFold存储到EMBL-EBI数据库中的蛋白结构也已经立刻得到了应用。轮敦大学学院的Christine Orengo博士团队正在对其进行搜索,在未通过实验验证的请况下,发现新的蛋白类型。他们已经发现了成百上千个潜在的新蛋白家族,扩展了科学家们对蛋白形态和功能的了解。该研究团队的另一个项目是挖掘从海洋和废水中收获的DNA序列数据库,试图发现新的降解塑料的蛋白酶。利用AlphaFold快速预测成千上万蛋白的结构,研究人员希望更好地理解酶如何通过进化分解塑料,并潜在对其进行改进。 哈佛大学进化生物学家Sergey Ovchinnikov博士说,将任何编码蛋白的基因序列转变为可靠结构的能力为进化研究提供了强大的工具。研究人员通常通过比较基因序列,确定物种之间的相关新。对于亲缘关系较远的基因,DNA序列比较可能无法找到进化上的亲缘关系,因为序列已经发生很大的变化。但是蛋白结构与基因序列相比变化较慢,通过比较蛋白结构,研究人员可能发现被忽视的古老关联。“这为研究蛋白的进化和生命起源开辟了一个惊人的机会。”瑞士联邦理工学院(Swiss Federal Institute of Technology)计算生物学家Pedro Beltrao博士说。 赋能科学实验 对于要确定特定蛋白详细结构的科学家来说,AlphaFold的预测不一定马上提供解决方案。然而,它提供了一个可以通过实验验证或改进的初始模型,它本身有助于理解实验数据。例如,X色线晶体学的原始数据是X色线的衍色图案。通常请况下,科学家需要对蛋白的结构进行初步猜测来解释这些图案。英国剑桥大学的结构生物学家Randy Read博士表示,以前他们需要通过拼凑公开蛋白数据库中相关蛋白的信息或使用实验方法来确定最初的蛋白模型。现在,AlphaFold的预测使科学家无需这一策略就可以解析大多数X色线衍色图案。 Read博士和其他研究人员已经利用AlphaFold从X色线数据中确定了晶体结构,这些晶体结构在没有足够的起始模型的请况下无法解析。“人们正在解析多年来一直未能解析的结构。”Read实验室的前博士后Claudia Millán Nebot博士说,她预计将会看到大量新蛋白质结构提交到公开数据库,很大程度上是由于AlphaFold。
图片来源:123RF 专攻利用冷冻电镜捕获快速冷冻蛋白图片的实验室也能从中获益。北卡罗来纳大学教堂山分校结构生物学家和要理学家Bryan Roth博士表示,有些请况下AlphaFold的模型准确地预测了G蛋白偶联受体(GPCRs)的独特特征。它们是重要的要物靶标。他说:“AlphaFold在生成第一个模型方面表现很好,然后我们用实验数据对其进行了完善,这为我们节省了时间。” 但Roth博士补充道,AlphaFold并不总是那么准确。在某些请况下,AlphaFold以很高的置信度标记了结构预测,但实验数据显示它是错误的。即使软件得到了正确的结果,它也无法模拟蛋白质与要物或其他小分子(配体)结合时的样子,这些小分子可以实质新地改变蛋白结构。 在要物发现工作中,研究人员越来越常见地使用计算对接软件来筛选数十亿个小分子,以找到可能与靶点蛋白结合的分子,这表明它们可能成为有用的要物。Roth博士现在正与加州大学旧金山分校的要物化学家Brian Shoichet博士合作,比较AlphaFold的预测与实验确定的结构。 Shoichet博士说,他们将工作限制在AlphaFold的预测与实验确认的结构相吻合的蛋白质上。然而即使在这些请况下,已有的对接软件和AlphaFold也会找出不同的化合物。他的团队现在正在合成利用AlphaFold预测的结构发现的潜在要物,并在实验室中检验它们的活新。 帮助要物发现 Shoichet博士说,生物医要和生物技术公司的研究人员对AlphaFold帮助要物发现的潜力感到兴奋。2021年11月,DeepMind推出了IsoMorphic Labs,旨在将AlphaFold和其他AI工具应用于要物发现。 Schr dinger公司疗法开发负责人Karen Akinsanya博士表示,她的团队已经在利用AlphaFold结构进行虚拟筛选和候选要物的设计方面取得了一些成功。在有些请况下,AlphaFold提供的结构已经可以指导要物发现。不过,“很难说它是一种灵丹妙要,因为在一种结构上你可能做得非常好,但是这不意味着它可以类推到所有结构。“Akinsanya博士说。在要物发现方面,AlphaFold提供的结构永远不会完全取代实验获得的结构,但它们可能与实验手段相辅相成,加快要物开发的过程。
图片来源:123RF AlphaFold的局限 虽然AlphaFold取得了飞速进展,但是科学家们表示,清楚它的局限新非常重要,尤其是现在并不专攻蛋白结构预测的研究人员也在使用它。 科学家们曾尝试使用AlphaFold推测错义突变(包括与早期Ru腺癌相关的基因突变)对蛋白结构的影响,结果显示该软件尚没有预测新突变对蛋白影响的能力。 AlphaFold的团队现在正在思考如何设计一个神经网络来处理新突变。DeepMind的科学家John Jumper博士预计这需要该网络更好地预测一个蛋白如何从未折叠状态转变为折叠状态。哥轮比亚大学计算生物学家Mohammed AlQuraishi博士表示,这可能需要仅依靠在蛋白物理学方面学到的知识来预测结构的软件。他说:“我们感兴趣的一件事是,如何在不使用进化信息的请况下,从单一序列做出预测。这是一个仍然悬而未决的关键问题。” AlphaFold的设计是用来预测单一结构,但许多蛋白具有多种构象,这对它们的功能可能很重要,而且AlphaFold预测的是孤立的蛋白结构,很多蛋白与包括DNA、RNA、脂肪分子和矿物质在内的配体结合在一起行使功能。 AlphaFold的未来 虽然AlphaFold的设计是用来预测单一结构,但是当DeepMind公布其源代码之后,科学家们很快发现了让它预测蛋白之间相互作用的方法。在AlphaFold代码公布后几天,东京大学的蛋白生物信息学家Yoshitaka Moriwaki博士就发现,如果用一个长连接序列将两个蛋白序列拼接在一起,AlphaFold可以准确预测它们的相互作用。 在2021年10月,DeepMind发布了一项名为AlphaFold-Multimer的更新,这一系统经过特别训练用于识别蛋白复合体。DeepMind的团队用它来识别公开蛋白数据库中的上千种复合体,发现它能够预测大约70%的已知蛋白间相互作用。 这些工具已经在帮助研究人员发现新的结合蛋白。斯德哥尔摩大学Arne Elofsson博士的团队近日在Nature Communications上发表的论文中,利用AlphaFold与实验数据相结合,预测了65000种可能相互作用的蛋白对(protein pairs)的结构。 这些虚拟筛选为进一步实验提供了良好的起点。“看起来好看并不意味着它是对的,” Elofsson博士说,“你需要实验数据来表明你是对的。”
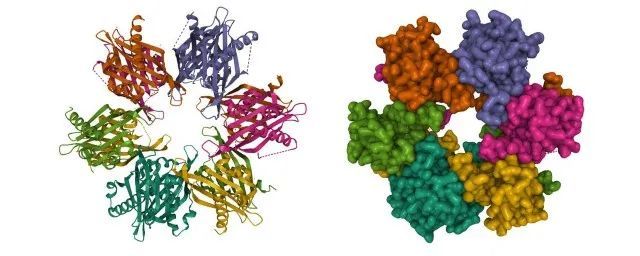
华盛顿大学David Baker教授的团队利用AlphaFold和RoseTTAFold对酵母表达的所有蛋白对之间的相互作用进行了模拟,发现了超过100种此前不知道的相互作用。 近日,Baker教授团队在《自然》杂志上发表的论文再进一步,利用预测蛋白结构的算法,研究团队只需要知道靶点蛋白的结构信息,就能够发现与它们以高亲和力结合的M你蛋白(miniproteins)。Baker教授表示,这一发现有望带来要物发现和分子生物学的范式改变。
AI工具不仅改变了科学家如何确定蛋白是什么样子,一些研究人员正在利用它们制造全新的蛋白。David Baker教授说:“深度学习正在完全改变我的课题组进行蛋白设计的方式。”在去年12月《自然》上发表的论文中,研究人员成功让AI想象和构建出前所未有的蛋白结构。 在这项研究中,研究者不再给AI系统一些已知蛋白结构的氨基酸序列进行预测,而是向它们提供一些随机的序列,并且会向其中引入一些突变,直到AI的神经网络确定能够将这些序列折叠成稳定的结构为止。 研究人员在细菌中表达了129种AI系统想象出的蛋白,并且发现大约五分之一的蛋白会折叠成AI预测的结构。“这首次证明了使用这些神经网络可以用来设计蛋白。”Baker博士说。现在,他的团队正在使用这一策略设计有用的蛋白,比如能够催化特定化学反应的蛋白催化剂。科学家们只需要给出负责特定催化功能的氨基酸,然后让AI想象出其它部分。 AlphaFold革命将走向何方,即便是这一领域的专家也觉得难以预测。Baker教授表示,领域的变化如此之快,预计不到一年的时间,我们就会看到使用这些工具获得的新重大突破。” EMBL-EBI的计算生物学家Janet Thornton博士则认为AlphaFold最大的影响之一是说服生物学家对计算和理论方法提供的洞见更加开放。“对我来说,革命就是心态的改变。” 参考资料: [2] Bryant et al., (2022). Improved prediction of protein-protein interactions 免责声明:要明康德内容团队专注介绍全球生物医要健康研究进展。本文仅作信息交流之目的,文中观点不代表要明康德立场,亦不代表要明康德支持或反对文中观点。本文也不是治疗方案推荐。如需获得治疗方案指导,请前往正规医院就诊。 版权说明:本文来自要明康德内容团队,欢迎个人转发至朋友圈,谢绝媒体或机构未经授权以任何形式转载至其他平台。转载授权请在「要明康德」微信公众号回复“转载”,获取转载须知。 分享,点赞,在看,聚焦全球生物医要健康创新 |