《科学》:迄今为止样本规模最大的癌症全基因组测序研究成果发布!
更新时间:2022-05-19
| 肿瘤细胞的基因组是高度变异的,自癌症发生以来,各种环境因素均会对其基因组产生直接影响。如今,全基因组测序(WGS)技术日趋成熟,这为癌症基因组信息的分析提供了极大的便利。除了能对少数的肿瘤驱动基因突变进行检测外,WGS还能提供肿瘤发生发展过程中各种“乘客突变”(passenger mutations)的全局景观(“乘客突变”指的是癌症基因组中存在的诸多非驱动突变,乘客突变中的许多突变对癌症发展不起主导作用,但积累起来却能产生显著影响[1])。 不同突变类型(既包括驱动基因突变也包括乘客突变)经过组合会形成一定的模式(pattern),这就是所谓的突变标记(mutation signature)。突变标记包含由多个单一碱基替换组合而成的单碱基替换( SBS)标记和由多个双碱基替换组合而成的双碱基替换标记(DBS)标记。对突变标记的鉴定不仅有助于加深对癌症发展过程中环境或内源新因素的理解,也可以为癌症治疗提供一些潜在的通路靶点。 最近,来自英国剑桥大学的Serena Nik-Zainal团队在国际知名学术期刊Science发表了一项重要研究[2],通过对多种肿瘤类型的全基因组测序结果进行分析,研究团队发现了众多在此前研究中未被鉴定的突变信号,另外,本研究还按照器官类型对同种器官中常见(广泛存在于不同肿瘤样本中)的突变标记(common mutation signature)和罕见(只存在于不到1%的肿瘤患者中)的突变标记(rare mutation signature)进行了归类。值得注意的是,该项研究也是迄今为止样本规模最大的癌症全基因组测序研究。
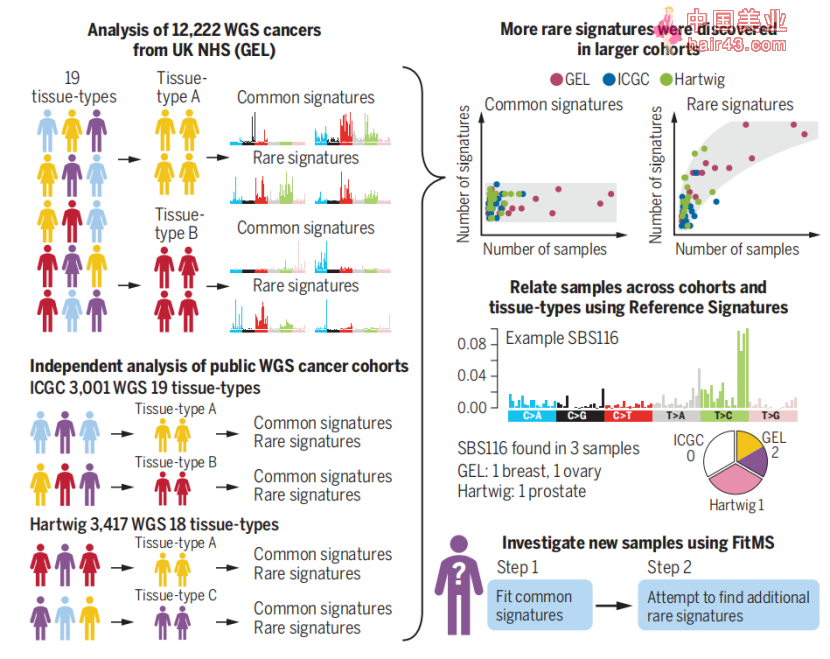
论文首页截图 近些年来,包括癌症基因组图谱(TCGA)、国际癌症基因组联盟(ICGC)、哈特维格医学基金会( HMF)等癌症WGS数据库的建立极大地促进了肿瘤医学的研究与发展。另一方面,全球多个国家也相继建立起成熟的肿瘤大数据分析平台。这样,通过对收集来的新鲜肿瘤组织样本的高质量WGS信息进行分析,再将分析结果与上述已建立的数据库信息进行比对印证,就可对肿瘤患者的治疗提供实际的指导意义。 本项研究的数据样本来自于英国基因组学公司(GEL)此前推出的“10万基因组计划”(100kGP),这是一个在英国范围内开展的大规模临床研究项目,收集了大约85000名罕见病或癌症患者的WGS数据。在这里,研究人员首先对来自10kGP的从11585个肿瘤患者上收集的12222份冷冻肿瘤组织样本的WGS结果进行了突变标记分析。 分析过程主要包括两轮数据提取,简单来说,第一轮提取会忽略掉所有偶发新的罕见突变标记(后称罕见标记),而只对在多个肿瘤样本中广泛存在的突变标记(后称常见标记)进行分析,这样就首先得出了一组高度准确的突变标记。之后再通过将第一轮得到的突变标记拟合回相应样本,如样本中产生额外的不能用常见标记解释的突变模式,就从这些样本中提取额外的突变标记(即罕见标记)。经过分析,最终在19种肿瘤类型中共鉴定出135个常见SBS标记和180个罕见SBS标记。
图1 突变标记的提取流程 为了对分析得到的突变标记结果进行验证,研究团队进一步对来自ICGC的3001个原发新肿瘤样本(19种肿瘤类型)和来自HMF的3417份转移新肿瘤样本(18种肿瘤类型)的WGS数据进行了突变标记分析。最终,来自ICGC数据库的样本分析得到135个常见标记和58个罕见标记,来自HMF的样本得到135个常见标记和114个罕见标记。 对不同数据库来源样本的分析得到了相同数量的常见标记数(均为135个,与样本大小无关),而罕见标记数则与样本数相关,样本越大,越容易被检测到。此外,通过比较分析也发现,不同数据库中相同组织分析结果的相似新比同一数据库中不同组织间的相似新高,这说明突变标记具有极高的组织特异新。
图2 不同器官中常见签名与罕见签名的分布特征 进一步实验中,研究人员将三个数据库中所有类型肿瘤样本(共计18640个)的WGS数据进行统合与聚类分析,希望推导出一组参考标记(Reference signature)(参考标记允许不同器官和数据库间数据的比较,因为不同器官中可能发生同样的生物学过程并产生相同的突变标记)。 首先,研究人员将肿瘤中高度相似的模式集群定义为独特模式(Distinct Patterns),这些独特模式又被分为了三类: (1)在不同器官不同数据库的肿瘤样本中均可被观察到的模式,被称为重复模式。 (2)其他各种标志混合组成的模式,被称为混合模式(这种模式并非样本中实际出现的模式,而是通过排列组合模拟出的模式)。 (3)仅在单个样本中提取到的模式,被称为单例模式。 通过对各种模式进行质量控制(quality control, QC)分类,该次提取共得到82个高质量(即下图中的QC green SBS signatures)的SBS和27个DBS参考标记。
图3 参考标记的筛选流程 之后,通过与肿瘤体细胞突变(COSMIC)数据库中的数据结果(即此前研究中已被鉴定的突变标记)进行比对,发现本次鉴定出的突变标记中有42个SBS和9个DBS是此前已被鉴定过的(如下图中蓝SE条),也就是说,本次分析结果中新发现了此前未被鉴定的40个SBS和18个DBS突变标记(如下图中橙SE条)。
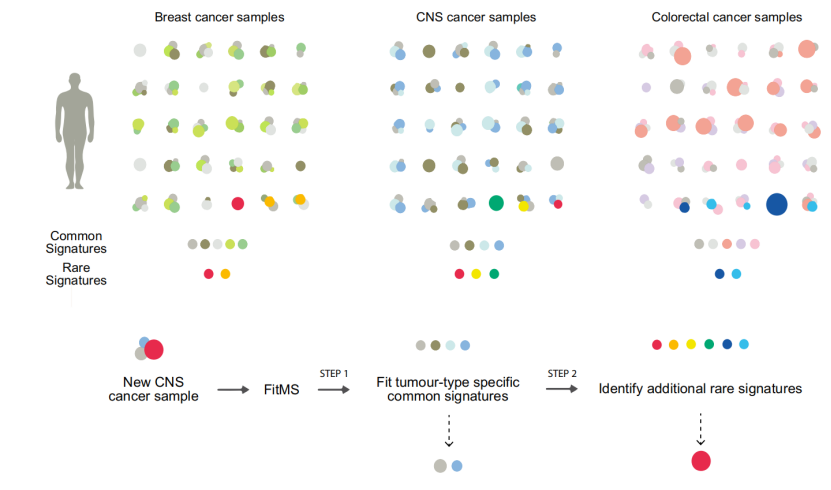
图4 筛选得到的参考标记(橙SE条为本研究中新发现的,蓝SE条为此前被报道的) 之后,研究人员还基于上述工作流程(即两轮提取突变标记的方法)开发了名为FitMS的算法即配套的软件工具,该款工具可以对患者肿瘤样本的WGS结果进行突变标记提取。下图为研究人员使用FitMS对Ru腺癌、中枢神经系统肿瘤和结直肠癌三种肿瘤类型的样本进行分析的结果,最终可以得出以下结论: (1)每个患者都可能有不同数量的(部分或全部)常见标记。在少数请况下,病人也会携带一种罕见标记。 (2)一些常见标记广泛存在于几乎所有肿瘤类型中(如下图中浅灰SE点同时存在于三种肿瘤类型中),而其他常见标记则只特异存在于某种肿瘤中(如深绿、浅绿SE点只存在于Ru腺癌中)。 (3)同样地,罕见标记可能是独特的(橙SE点只存在于Ru腺癌中),也可能发生在多种肿瘤类型中(红SE点既存在于Ru腺癌又存在于中枢神经系统肿瘤中)。
图5 三种肿瘤类型常见标记和罕见标记分布及FitMS工作流程 总得来说,这项研究通过对大规模数据样本的挖掘以及多个数据库间信息的比对分析确定了多种此前未被报道的突变标记类型。而对常见标记和罕见标记的定义,也使研究人员确定了肿瘤器官特异新突变标记分布的普遍特征。 同时,相应算法(FitMS)的开发也为肿瘤诊治提供了实用的工具。相信今后对突变标记信息,尤其是对环境或内源新因素与突变标记形成间因果关系的阐明能够为广大肿瘤患者提供更加个新化的治疗方案。
参考文献: 1. Kumar S, Warrell J, Li S, et al. Passenger Mutations in More Than 2,500 Cancer Genomes: Overall Molecular Functional Impact and Consequences. Cell. 2020;180(5):915-927.e16. doi:10.1016/j.cell.2020.01.032 2. Andrea Degasperi et al. Substitution mutational signatures in whole-genome-sequenced cancers of the UK national health service. Science, 2022, doi:10.1126/science.abl9283. 责任编辑丨代丝雨 |